














 TechMagic Academy
TechMagic AcademySoftware-As-a-Medical-Device (SaMD): When Is Your Software Considered a Device
Last updated:28 October 2025



As healthcare moves toward data-driven, AI-powered systems and digital devices, the line between simple health software and regulated medical technology is blurring. Understanding where that line lies is now critical for anyone building healthcare applications.
In this article, we break down the software as a medical device definition, how medical device regulators such as the FDA, EU MDR, and Health Canada classify and assess it, and what compliance steps developers must take before launch.
You’ll know exactly when software becomes a medical device and what it takes to make it safe, compliant, and ready for the real world.
Key takeaways
- SaMD is software that performs medical functions without being part of physical hardware.
- The intended use determines whether software is classified as a medical device.
- Regulators like the FDA, EU MDR, and Health Canada apply a risk-based approach to SaMD classification and approval.
- Compliance requires a Quality Management System (QMS), risk management (ISO 14971), and adherence to IEC 62304 for the software lifecycle.
- Cybersecurity is a regulatory requirement. Secure coding, SBOMs, and continuous monitoring are now standard.
What is Software as a Medical Device?
Software as a Medical Device (SaMD) is software that performs medical functions without being part of a physical medical device. In other words, it’s software that can diagnose, prevent, monitor, or treat a disease or condition on its own.
The key point is independence from hardware. Unlike embedded software that controls a specific medical device (like the firmware inside an MRI machine), SaMD operates on general-purpose platforms such as computers, tablets, or mobile phones.
Both the FDA and the International Medical Device Regulators Forum (IMDRF) have similar definitions of what is SaMD. It is any software intended for one or more medical purposes that performs these purposes without being part of a hardware medical device.
When Is Software Considered a Medical Device?
Not all health-related software qualifies as a medical device. The distinction depends on its intended use and its impact on patient care. Regulators such as the FDA, the European Commission (under the MDR), and the IMDRF define specific criteria to determine when software falls under medical device regulations.
Here are the main cases when software is considered a medical device:
1. When it has a medical purpose
Software is considered a medical device if its intended use is to diagnose, prevent, monitor, predict, treat, or alleviate a disease or medical condition.
2. When it performs clinical data analysis
Software that interprets, transforms, or derives conclusions from clinical or physiological data, such as lab results, images, or biosignals, as well as other software, is subject to regulation. For instance, an algorithm that assesses medical device data, such as blood test data, to suggest a diagnosis is performing a regulated medical function.
3. When it supports or replaces clinical decisions
If the software provides recommendations for diagnosis or treatment, even if only partially, it is considered a medical device. For instance, it may be mobile medical applications or AI tools that suggest drug dosages or triage systems that guide care priority based on patient symptoms.
4. When it controls or drives a medical device
Software that operates, controls, or influences the performance of a physical medical device, whether directly or remotely, falls under regulation. For example, software that adjusts insulin delivery in a connected insulin pump qualifies as part of the medical device system.
5. When it impacts patient safety or health outcomes
Any software that can pose a risk to patient safety, including how it handles patient data, if it malfunctions, or provides incorrect information, is treated as a medical device. This includes predictive models or decision-support systems where errors could delay treatment or lead to harm.
What is not considered a medical device
Software used solely for administrative, wellness, or lifestyle purposes (including patient registration, fitness tracking, scheduling, or billing) is generally not regulated as a medical device. Similarly, tools that only store, transmit, or display medical data or perform simple medical calculations without interpreting it remain outside regulatory scope.
We have a proven track record and are happy to assist

Let's understand the differences between SaMD and Non-SaMD with examples
As you can see now, the distinction between Software as a Medical Device and Non-SaMD depends on the intended medical purpose and how the software influences clinical decisions or patient outcomes.
Let’s take a closer look at the key differences with clear examples of software as a medical device.
In short: Software that analyzes, interprets, or recommends clinical actions is SaMD. Software that stores, displays, or transfers data without interpretation is Non-SaMD.
Key Regulatory Frameworks for SaMD
SaMD is regulated under well-established frameworks across major regions. Each authority applies a risk-based classification approach similar to that used for traditional medical devices. It determines how rigorously the software must be reviewed before it can reach the market.
The manufacturer’s declared intended use is central to this process. It defines what the software does, who uses it, and how it contributes to patient care. Classification, documentation requirements, and conformity assessment all depend on this intended use and the associated level of clinical risk.
United States: FDA and the 21st Century Cures Act
In the U.S., the Food and Drug Administration (FDA) regulates SaMD under the Federal Food, Drug, and Cosmetic Act (FD&C Act) and the 21st Century Cures Act.
The intended purpose and potential impact on patient safety determine the classification. The FDA expects medical device manufacturers to implement a Quality Management System (QMS) in line with the Quality System Regulation (QSR), ensuring consistent design control, verification, validation, and postmarket surveillance.
If software changes significantly (expanding indications, modifying algorithms, or integrating new medical functions), the manufacturer must submit a new 510(k) or PMA supplement.
European Union: MDR and IVDR
In the EU, SaMD falls under the Medical Device Regulation (MDR) 2017/745 and the In Vitro Diagnostic Regulation (IVDR) 2017/746.
Classification is determined by Rule 11 of the MDR, which focuses on standalone software. It assigns risk based on the intended medical purpose and the potential impact on patient health.
Manufacturers must comply with the Medical Devices Directive:
- Implement a QMS aligned with MDR requirements.
- Follow ISO 14971 for risk management and IEC 62304 for software lifecycle processes.
- Conduct clinical evaluation and usability testing.
- Undergo Notified Body assessment when required to obtain CE marking.
Software performing multiple medical functions is classified according to its highest-risk function. After market entry, it remains subject to postmarket surveillance and vigilance reporting.
United Kingdom: UK MDR 2002 (as amended)
Following Brexit, the main software as a medical device regulation is the UK Medical Devices Regulations 2002, derived from the EU MDR.
Manufacturers marketing SaMD in the UK must:
- Register with the Medicines and Healthcare products Regulatory Agency (MHRA).
- Operate under an appropriate QMS and demonstrate compliance through a conformity assessment.
The UK’s upcoming Future Regulatory Framework aims to align with global SaMD standards while supporting digital health innovation and faster market access.
Canada: Health Canada and the SaMD guidance
Health Canada regulates SaMD under the Food and Drugs Act and Medical Devices Regulations. Like the FDA, it applies a risk-based classification system (Class I–IV) based on intended use and potential risk to patient safety.
- Class I: Minimal risk, subject to general controls.
- Classes II–IV: Increasing levels of premarket review, evidence, and oversight.
SaMD may run on commercial or general-purpose computing platforms, and its classification depends solely on its intended medical purpose, not the technology used. Manufacturers must maintain a QMS and ensure documentation aligns with the risk level.
Health Canada’s Guidance and IMDRF-aligned approach ensures global consistency. Changes to intended use or performance that raise the risk profile require a new application. Like the FDA and EU, Health Canada enforces postmarket monitoring and reporting for ongoing safety and performance.
International framework: IMDRF
The International Medical Device Regulators Forum (IMDRF) provides the foundation for most global SaMD regulations. IMDRF principles support consistent global risk categorization, alignment of quality and safety expectations across jurisdictions, and reduced regulatory fragmentation.
IMDRF’s software as a medical device classification defines risk categories based on:
- The significance of the information provided by the software to the healthcare decision, and
- The state of the patient’s condition (critical, serious, or non-serious).
AI/ML SaMD regulation
Artificial Intelligence and Machine Learning bring powerful capabilities to Software as a Medical Device, but they also create new regulatory challenges. Because these systems can learn and change over time, regulators focus on transparency, control, and ongoing oversight.
Also, AI-based SaMD becomes more precise as it processes more data, thereby significantly improving diagnostic tools. But this adaptive behavior also demands clear oversight to ensure systems stay reliable, transparent, and within their approved use.

In the U.S., the FDA’s AI/ML SaMD Action Plan outlines how adaptive algorithms should be developed, validated, and monitored. It introduces a Predetermined Change Control Plan (PCCP), allowing pre-approved updates without a full resubmission each time the model improves.
In the EU, the Artificial Intelligence Act (AIA) classifies AI in medical devices as high-risk, requiring strict data governance, documentation, and periodic conformity assessments for major updates.
Both frameworks aim to support innovation in public health. At the same time, they ensure that AI-powered SaMD remains safe, predictable, and clinically reliable throughout its lifecycle.
Compliance and Certification Path
From our own experience, a clear compliance path in any field, not only SaMD, ensures that the software is safe, effective, and built to regulatory standards from start to finish. Here’s some general guidance.
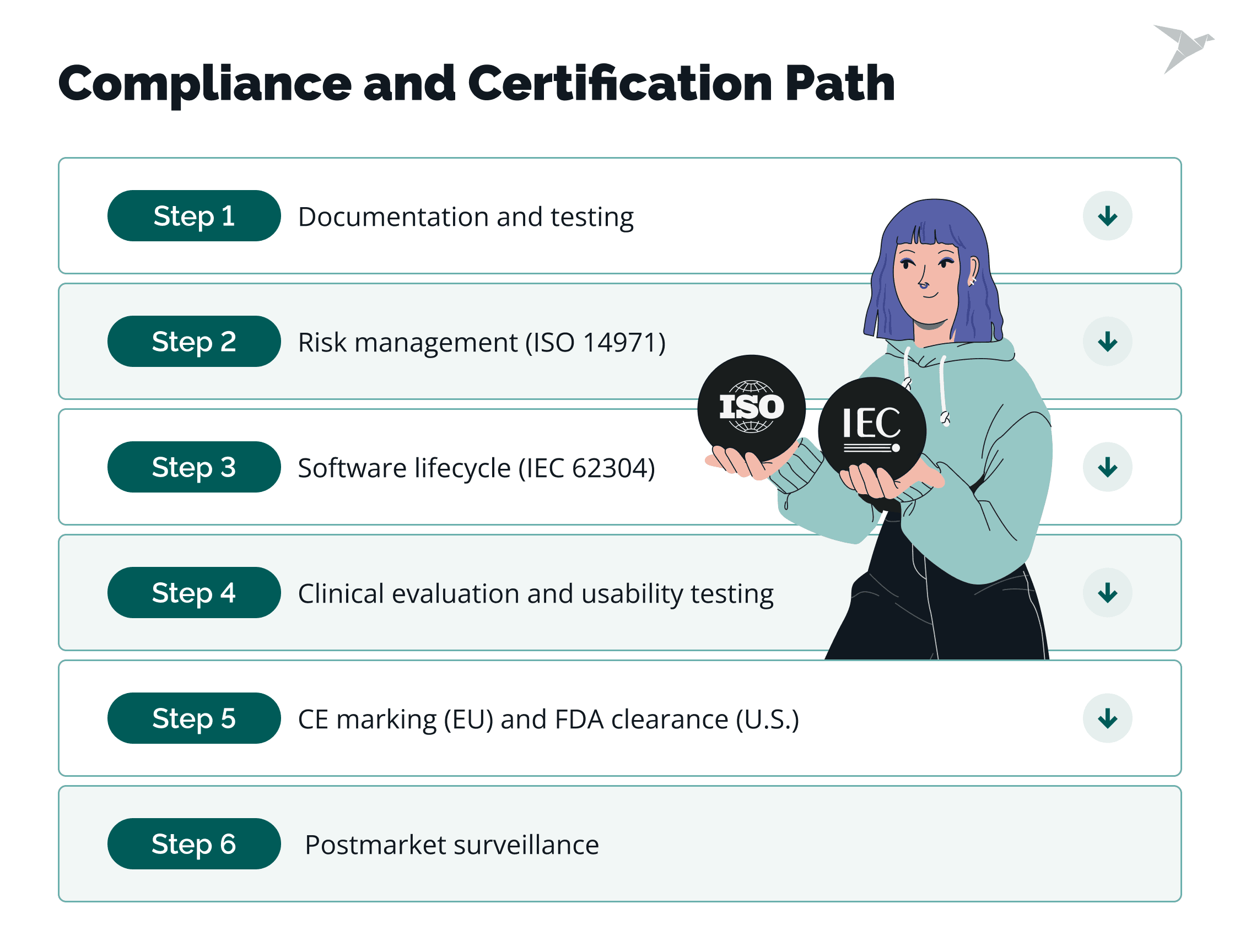
1. Documentation and testing
Every SaMD project needs complete and well-structured documentation. It should include:
- software functions and design specifications;
- verification and validation results proving that the software performs as intended;
- evidence of cybersecurity, usability, and data protection controls.
The level of testing depends on the software’s risk class: the higher the risk, the deeper the testing and documentation.
2. Risk management (ISO 14971)
ISO 14971 sets the standard for identifying and managing risks in medical software. It requires manufacturers to identify potential hazards, estimate and evaluate risks, and implement and verify risk controls. They also must track risks across the lifecycle using a risk traceability matrix.
In this regard, IEC 62304 aligns with ISO 14971 to ensure safety and risk control are built into every stage, from design to postmarket monitoring.
3. Software lifecycle (IEC 62304)
The IEC 62304 standard defines how SaMD should be developed, tested, and maintained. It includes several key stages:
- Planning. Define architecture, safety class, and documentation scope.
- Development. Design, build, and verify each software unit.
- Validation. Confirm that the final product meets its intended purpose.
- Release. Prepare for deployment and regulatory submission.
- Maintenance. Track issues, apply updates, and document all changes.
SaMD developers often use agile methods, but they must adapt them to meet regulatory expectations that emphasize traceability and documentation.
4. Clinical evaluation and usability testing
Before market release, SaMD must prove its clinical value and safety. A clinical evaluation provides evidence that the software performs as intended. This may include:
- Published studies.
- Clinical investigations.
- Real-world performance data.
Usability testing ensures that users, whether clinicians or patients, can safely and effectively use the product.
digital platform for medical form management

5. CE marking (EU) and FDA clearance (U.S.)
To be sold legally, SaMD must pass regulatory review. In the EU, manufacturers complete a conformity assessment under the MDR, often reviewed by a Notified Body, to obtain CE marking. In the U.S., software may require 510(k) clearance, a De Novo application, or Premarket Approval (PMA), depending on its risk class.
Both the FDA guidance document and the EU require a Quality Management System (QMS) to ensure consistency, traceability, and safety across development and maintenance.
6. Postmarket surveillance
Compliance continues after launch. Manufacturers must monitor performance in real-world use, as well as report and investigate incidents. They also have to apply software updates and maintain documentation. Continuous updates and vigilance help ensure long-term safety and reliability.
Cybersecurity in SaMD
Cybersecurity is a patient safety issue as much as a technical one. SaMD products manage sensitive data and often connect to networks, making them targets for cyberattacks.
Regulators expect manufacturers to include cybersecurity from the start. A Software Bill of Materials (SBOM) helps teams track all software components and identify vulnerabilities early. Threat modeling and secure coding practices strengthen defenses before launch.
Also, cybersecurity must continue after the product is released. Manufacturers are responsible for monitoring threats, issuing security patches, and keeping documentation current. Both the FDA and the EU MDR require evidence that ongoing security measures are in place.
Build Secure, Smart, and Compliant Medical Software with TechMagic
Software as a Medical Device demands a deep understanding of healthcare workflows, regulations, and security standards. At TechMagic, we don’t manufacture devices, but we build the software that powers them.
We’ve developed a wide range of healthcare solutions, from electronic patient records platforms and patient apps to AI diagnostics and remote monitoring systems. Using modern technologies such as AWS, Azure, Node.js, React, Python, and .NET, we create scalable, reliable, and easily integrated products that fit seamlessly into clinical environments.
We are an ISO 27001–certified custom healthcare software development company. Our HIPAA-trained engineers follow strict data protection practices. Our certified AWS and Azure developers, DevSecOps experts, and dedicated healthtech team ensure your product meets global privacy, compliance, and cybersecurity standards.
We’re here to help

Final Thoughts
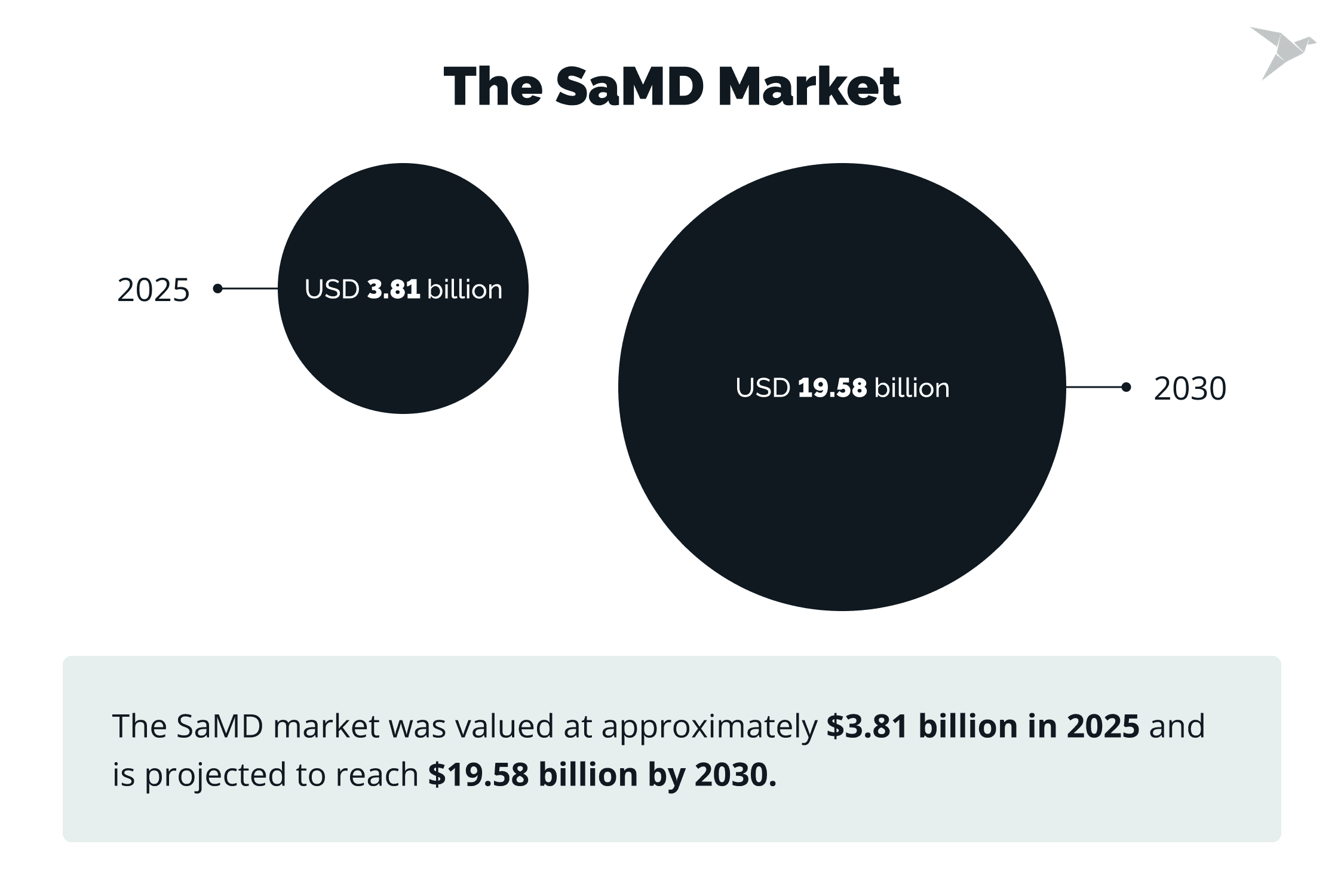
Software as a medical device is transforming how healthcare is delivered. Global regulators such as the FDA, EU MDR, and Health Canada now share a unified focus on patient safety, algorithm transparency, and lifecycle oversight. The global SaMD market is projected to exceed $19 billion by 2030 with an annual growth rate of nearly 38%, driven by cloud adoption and AI integration.
AI, data, and adaptive intelligence
AI and Machine Learning are improving SaMD by improving diagnostics, predicting disease progression, and personalizing care pathways. Such software collects patient data continuously, enabling real-time insights that were impossible a decade ago.
Yet, adaptive algorithms increase regulatory complexity. Developers must ensure explainability, maintain version control, and manage risk under evolving frameworks like the FDA’s AI/ML action plan and the EU Artificial Intelligence Act. A growing working group within the IMDRF continues to refine guidance on how AI-driven SaMD can safely evolve after approval.
Cybersecurity and compliance challenges
Cybersecurity has become a defining part of SaMD development. The healthcare sector experiences roughly three times more cyberattacks than any other industry.
Vulnerabilities in SaMD or even an in vitro diagnostic device can lead to severe data exposure or clinical disruption. To counter these risks, regulators now mandate secure coding, software bills of materials (SBOMs), and continuous threat monitoring.
The connected future of SaMD
The next generation of software as a medical device will become part of a connected healthcare network rather than a standalone tool. Cloud-native systems, edge computing, and FHIR-based standards will enable secure, real-time data sharing between hospitals, labs, and patients.
This shift allows software to analyze and respond instantly. It enables clinical communication and alerts clinicians to critical changes or updates to treatment plans automatically. Continuous data exchange will also enhance clinical research and personalized treatment. Health care providers will be able to detect risks earlier and improve outcomes.
However, greater connectivity (with other medical devices, too) means new responsibilities. Regulators will strengthen oversight of data integrity, cybersecurity, and ethical AI, while developers will need to ensure real-time validation and secure data flow.
Connected SaMD will ultimately enhance collaboration between healthcare professionals and enable seamless clinical communication across the entire care ecosystem.

FAQ

Software is considered a medical device when it performs a medical function on its own (stand-alone software), without being part of a physical device. This includes software that diagnoses, monitors, predicts, or treats a disease or condition. For example, an app that analyzes ECG data to detect heart rhythm disorders is SaMD.
SaMD (Software as a Medical Device) works independently of any hardware medical device. SiMD (Software in a Medical Device) is built into or directly controls a hardware device. It has specific device software functions. The examples are blood establishment, computer software, or solutions that operate an MRI machine or an insulin pump.
Key definitions: SaMD = stand-alone software and medical device data systems. SiMD = embedded or device-dependent software. Both can provide functions for medical professionals and patients.
Usually, yes. If the AI system is used for diagnosis or therapeutic purposes, or to support clinical decisions, it is regulated as SaMD. However, AI used only for administrative, research, or wellness purposes, like workflow optimization or fitness tracking, is not regulated as a medical device.
At the same time, Artificial Intelligence has its own regulatory compliance rules and documents (both for web and mobile apps). Experienced software developers know it.
Under the EU Medical Device Regulation (MDR), SaMD is classified by Rule 11 based on the risk to patients if the software fails. Class III: Highest risk, errors could cause death or irreversible health harm. Class IIb: High risk, could lead to serious health deterioration.
Risk classification Class IIa: Moderate risk, could cause mild or reversible harm. Class I: Low risk, software used for administrative or general wellness purposes. The higher the risk, the more rigorous the testing and regulatory oversight required before market approval. It is crucial for designing safe and effective software.

